Topics:
Topics:
Researchers at the RIKEN Center for Sustainable Resource Science (CSRS) in Japan have developed a new computational mass-spectrometry system for identifying metabolomes—entire sets of metabolites for different living organisms. When the new method was tested on select tissues from 12 plants species, it was able to note over a thousand metabolites. Among them were dozens that had never been found before, including those with antibiotic and anti-cancer potential.

This study made the cover of Nature Methods!
The common pain reliever aspirin (acetylsalicylic acid) was first made in the 19th century, and is famously derived from willow bark extract, a medicine that was described in clay tablets thousands of years ago. After a new method of synthesis was discovered, and after it had been used around the world for almost 70 years, scientists were finally able to understand how it works. This was a long historical process, and while plants remain an almost infinite resource for drug discovery and biotechnology, thousands of years is no longer an acceptable time frame.
Why does it take so long?
The biggest problem is that there are millions of plant species and each has its own metabolome—the set of all products of the plant’s metabolism. Currently, we only know about 5% of all these natural products. Although mass spectrometry can identify plant metabolites, it only works for determining if a sample contains a given molecule. Searching for as-yet-unknown metabolites is another story.
Computational mass spectrometry is a growing research field that focuses on finding previously unknown metabolites and predicting their functions. The field has established metabolome databases and repositories, which facilitate global identification of human, plant, and microbiota metabolomes. Led by Hiroshi Tsugawa and Kazuki Saito, a team at CSRS has spent several years developing a system that can quickly identify large numbers of plant metabolites, including those that have not been identified before.
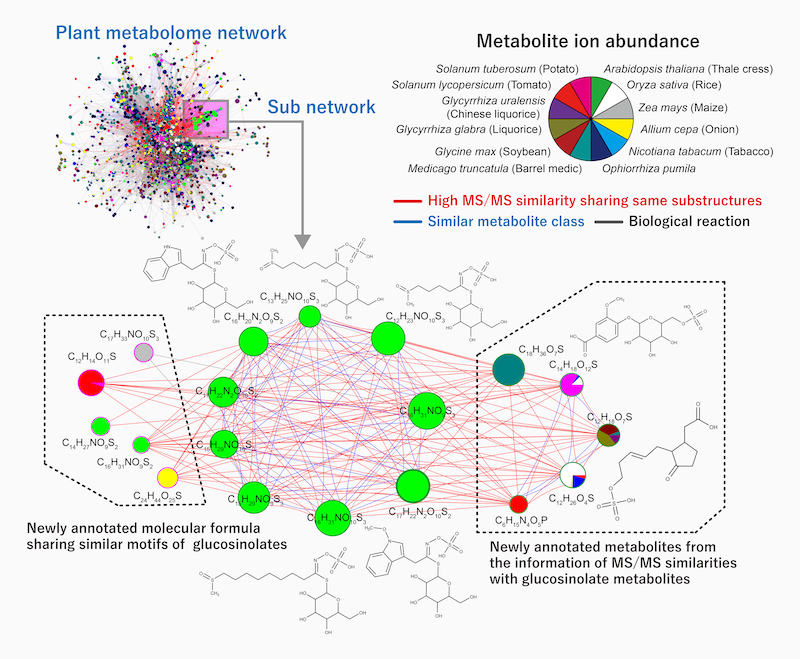
The results of the experiment. Within a subnetwork of a plant’s metabolome, the new compMS system was able to find novel metabolites that highly resemble glucosinolate metabolites in substructure, and well as those that share a similar motif with glucosinolates. Glucosinolates are compounds found in plants such as mustard, cabbage, and horseradish.
As Tsugawa explains, “while no software can comprehensively identify all the metabolites in a living organism, our program incorporates new techniques in computational mass spectrometry and provides 10 times the coverage of previous methods.” In tests, while mass spectrometry-based methods only noted about one hundred metabolites, the team’s new system was able to find more than one thousand.
The new computational technique relies on several new algorithms that compare the mass spectrometry outputs from plants that are labeled with carbon-13 with those that are not. The algorithms can predict the molecular formula of the metabolites and classify them by type. They can also predict the substructure of unknown metabolites, and based on similarities in structure, link them to known metabolites, which can help predict their functions.
Being able to find unknown metabolites is a key selling point for the new software. In particular, the system was able to characterize a class of antibiotics (benzoxazinoids) in rice and maize as well as a class with anti-inflammatory and antibacterial properties (glycoalkaloids) in the common onion, tomato, and potato. It was also able to identify two classes of anti-cancer metabolites, one (triterpene saponins) in soy beans and licorice, and the other (beta-carboline alkaloid) in a plant from the coffee family.

Triterpene saponins are metabolites that have anti-cancer properties. The new computational mass spectrometry system found novel metabolites of this class in beans and licorice.
In addition to facilitating the screening of plant-specialized metabolomes, the new process will speed up the discovery of natural products that could be used in medicines, and also increase understanding of plant physiology in general.
Tsugawa notes that use of this new method is not limited to plants. “I believe that computationally decoding metabolomic mass spectrometry data is linked to a deeper understanding of all metabolisms. Our next goal is to improve this methodology to facilitate global identification of human and microbiota metabolomes as well. Newly found metabolites can then be further investigated via genomics, transcriptomics, and proteomics.” ✅
Researchers at the RIKEN Center for Sustainable Resource Science (CSRS) in Japan have developed a new computational mass-spectrometry system for identifying metabolomes—entire sets of metabolites for different living organisms. When the new method was tested on select tissues from 12 plants species, it was able to note over a thousand metabolites. Among them were dozens that had never been found before, including those with antibiotic and anti-cancer potential.
This study made the cover of Nature Methods!
The common pain reliever aspirin (acetylsalicylic acid) was first made in the 19th century, and is famously derived from willow bark extract, a medicine that was described in clay tablets thousands of years ago. After a new method of synthesis was discovered, and after it had been used around the world for almost 70 years, scientists were finally able to understand how it works. This was a long historical process, and while plants remain an almost infinite resource for drug discovery and biotechnology, thousands of years is no longer an acceptable time frame.
Why does it take so long?
The biggest problem is that there are millions of plant species and each has its own metabolome—the set of all products of the plant’s metabolism. Currently, we only know about 5% of all these natural products. Although mass spectrometry can identify plant metabolites, it only works for determining if a sample contains a given molecule. Searching for as-yet-unknown metabolites is another story.
Computational mass spectrometry is a growing research field that focuses on finding previously unknown metabolites and predicting their functions. The field has established metabolome databases and repositories, which facilitate global identification of human, plant, and microbiota metabolomes. Led by Hiroshi Tsugawa and Kazuki Saito, a team at CSRS has spent several years developing a system that can quickly identify large numbers of plant metabolites, including those that have not been identified before.
The results of the experiment. Within a subnetwork of a plant’s metabolome, the new compMS system was able to find novel metabolites that highly resemble glucosinolate metabolites in substructure, and well as those that share a similar motif with glucosinolates. Glucosinolates are compounds found in plants such as mustard, cabbage, and horseradish.
As Tsugawa explains, “while no software can comprehensively identify all the metabolites in a living organism, our program incorporates new techniques in computational mass spectrometry and provides 10 times the coverage of previous methods.” In tests, while mass spectrometry-based methods only noted about one hundred metabolites, the team’s new system was able to find more than one thousand.
The new computational technique relies on several new algorithms that compare the mass spectrometry outputs from plants that are labeled with carbon-13 with those that are not. The algorithms can predict the molecular formula of the metabolites and classify them by type. They can also predict the substructure of unknown metabolites, and based on similarities in structure, link them to known metabolites, which can help predict their functions.
Being able to find unknown metabolites is a key selling point for the new software. In particular, the system was able to characterize a class of antibiotics (benzoxazinoids) in rice and maize as well as a class with anti-inflammatory and antibacterial properties (glycoalkaloids) in the common onion, tomato, and potato. It was also able to identify two classes of anti-cancer metabolites, one (triterpene saponins) in soy beans and licorice, and the other (beta-carboline alkaloid) in a plant from the coffee family.
Triterpene saponins are metabolites that have anti-cancer properties. The new computational mass spectrometry system found novel metabolites of this class in beans and licorice.
In addition to facilitating the screening of plant-specialized metabolomes, the new process will speed up the discovery of natural products that could be used in medicines, and also increase understanding of plant physiology in general.
Tsugawa notes that use of this new method is not limited to plants. “I believe that computationally decoding metabolomic mass spectrometry data is linked to a deeper understanding of all metabolisms. Our next goal is to improve this methodology to facilitate global identification of human and microbiota metabolomes as well. Newly found metabolites can then be further investigated via genomics, transcriptomics, and proteomics.” ✅
Further reading
Tsugawa et al. (2019) A cheminformatics approach to characterize metabolomes in stable isotope-labeled organisms. Nature Methods. doi: 10.1038/s41592-019-0358-2