Researchers at the RIKEN Center for Brain Science (CBS) in Japan have discovered that the protein α-endosulfine (ENSA) is involved in the development of Alzheimer’s disease. Studies in mice showed that eliminating this protein entirely or using drugs to block its function reduced physical changes in the brain associated with the disease and improved memory. Drug therapy that aims to block ENSA activity could be a more effective treatment than what is currently available, as well as being cheaper. This study was published in the scientific journal Molecular Psychiatry.
Topics:
Topics:
The hallmark of Alzheimer’s disease in the brain is the accumulation of amyloid β peptide (Aβ). For years, researchers have been trying to determine how and why this happens. Takaomi Saido and his team at RIKEN CBS have developed a mouse model of the disease that shows both Aβ accumulation and memory deficits similar to what is seen in humans. Using this model mouse, they have already discovered a series of events in the brain that lead to the formation of Aβ plaques. Key among them is reduced levels of the enzyme neprilysin, which itself is caused by reduced levels of the hormone somatostatin. Levels of both neprilysin and somatostatin go down as we age, which can explain why Alzheimer’s disease usually strikes older people.
The new study focused on treating Alzheimer’s disease in mice by figuring out how somatostatin controls neprilysin levels in the brain. According to first author Naoto Watamura, “the first step in this process was actually the most difficult because we had to develop an in vitro system that could screen for neprilsyin regulators in conditioned medium generated by hippocampal neurons.” Once they accomplished this, they were able to identify ENSA as the regulator. Testing showed that ENSA reduced neprilysin activity and that it rose to abnormally high levels in the brains of mice that lacked somatostatin. This means that somatostatin normally keeps ENSA in check, which in turn keeps neprilsyin levels high, allowing Aβ to be destroyed before it accumulates.
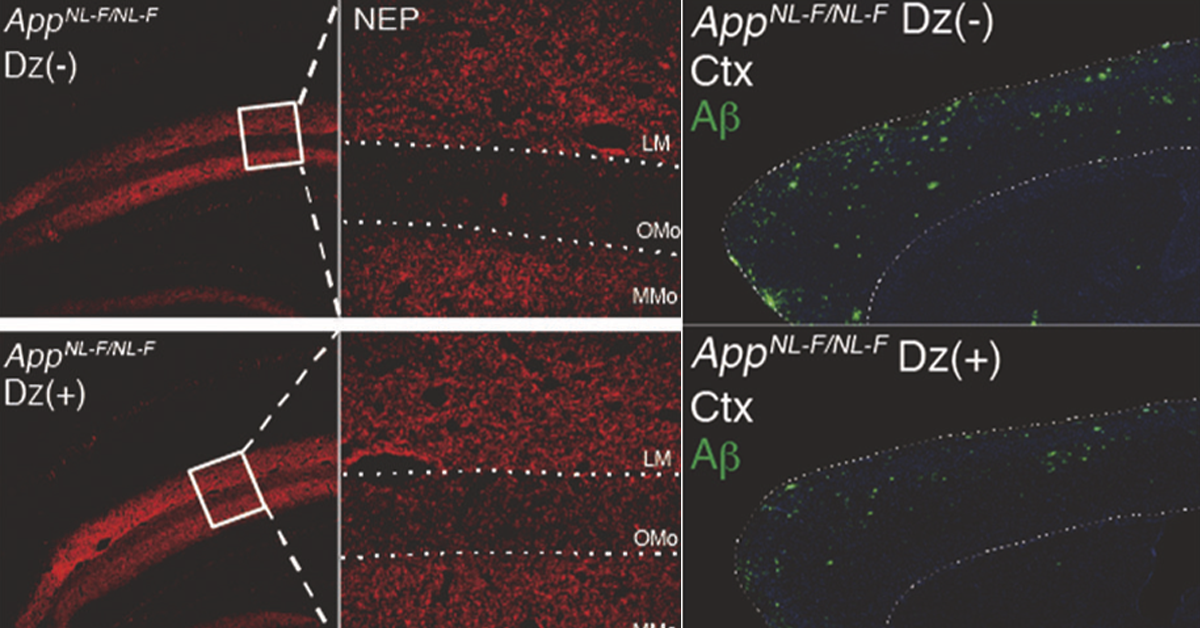
Treatment with diazoxide reduced Aβ pathology in mice with Alzheimer’s disease. Neprilysin (red) and Aβ (green) levels in the hippocampus of Alzheimer’s disease model mice. Top: control. Bottom: after being fed diazoxide. After diazoxide treatment, neprilysin levels increased and Aβ levels decreased. Mice also performed better on memory tests (not shown here).
Next the team focused on ENSA in living animals. Using CRISPR technology, they created ENSA knockout mice and then bred them with the Alzheimer’s disease model mice. Aβ accumulation in these new mice was much lower than in the original model mice, indicating that abnormally high levels of ENSA could be an as yet unidentified symptom or biomarker of Alzheimer’s disease. This was confirmed when the researchers detected high level of ENSA in the model mice and in the brains of people with Alzheimer’s disease.
What exactly is ENSA doing in the brain? Tests showed that ENSA blocks a potassium channel in the hippocampus, a part of the brain needed for making and recalling memories. “Because we got the same results from blocking the KATP channel as we did from the ENSA knockout mice,” says Watamura, “we reasoned that helping the channel stay open would combat the excess ENSA that we observed in Alzheimer’s disease.” To test this theory, the researchers fed the model mice with diazoxide — a drug that activates the KATP channel — and tested their memory. They found that while the untreated Alzheimer’s disease model mice exhibited their characteristically poor memory, the treated model mice performed just as well as normal mice. A look at the brains of the treated mice showed that they lacked the hallmark Aβ plaques.
“Our findings point directly to a potential way of preventing and treating Alzheimer’s disease,” says Watamura. “On top of that, compared with Aβ-targeting immunotherapy, such as the drug aducanumab, which was recently approved by the FDA, synthetic agonists for the KATP channel are less expensive and would be more acceptable to aging societies around the world.” ?
Further reading
Watamura et al (2021) Somatostatin-evoked Aβ catabolism in the brain: Mechanistic involvement of α-endosulfine-KATP channel pathway. Mol Psychiatry. doi: 10.1038/s41380-021-01368-8.
Further reading
Watamura et al (2021) Somatostatin-evoked Aβ catabolism in the brain: Mechanistic involvement of α-endosulfine-KATP channel pathway. Mol Psychiatry. doi: 10.1038/s41380-021-01368-8.
")








